

蛋白质结构相比于序列往往被认为更加具有信息量,因为其直接决定了蛋白质的功能。
而随着AlphaFold2带来的巨大突破,大量的预测结构被发布出来供人研究使用,如何利用这些蛋白质结构来训练强大且通用的表征模型是一个值得研究的方向。
西湖大学的研究人员利用Foldseek来处理蛋白质结构,将其编码成一维的离散token,并与传统的氨基酸进行结合,形成了结构感知词表(Structure-aware Vocabulary),以此将结构信息嵌入到模型输入中,增强模型的表征能力。

论文地址:https://www.biorxiv.org/content/10.1101/2023.10.01.560349v4
Github地址:https://github.com/westlake-repl/SaProt
在预训练上,本文使用了目前最多的蛋白质结构(大约4000万),在64张A100上训练了3个月,最终开源了具备650M参数量的模型SaProt(同时包括了35M的版本)。实验结果表明SaProt各种蛋白质任务上都要好于之前的序列和结构模型。
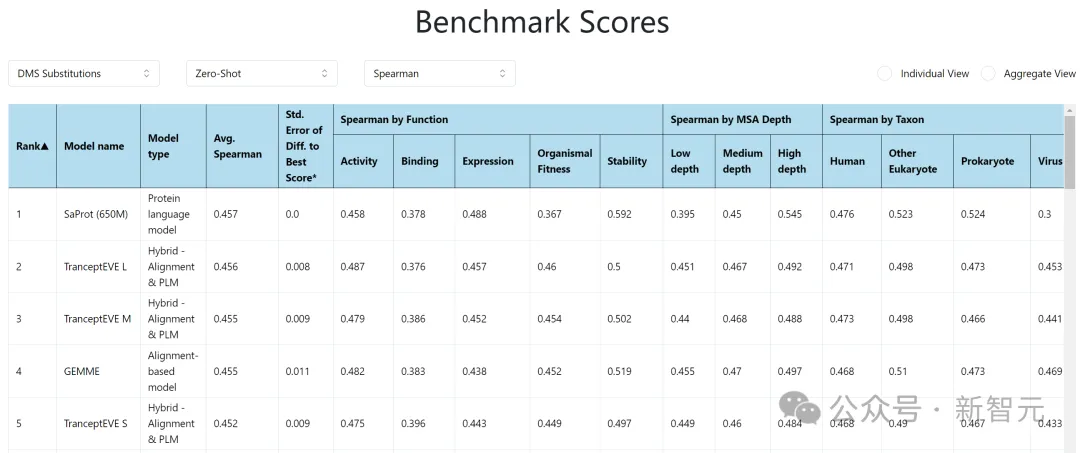 ProteinGym:https://proteingym.org/benchmarks
ProteinGym:https://proteingym.org/benchmarks
方法
该研究利用Foldseek将蛋白质进行编码,生成了一维的3Di结构序列(使用了Foldseek的结构词表,每种3Di token代表不同的局部结构),这样的结构序列与氨基酸序列是等长的。
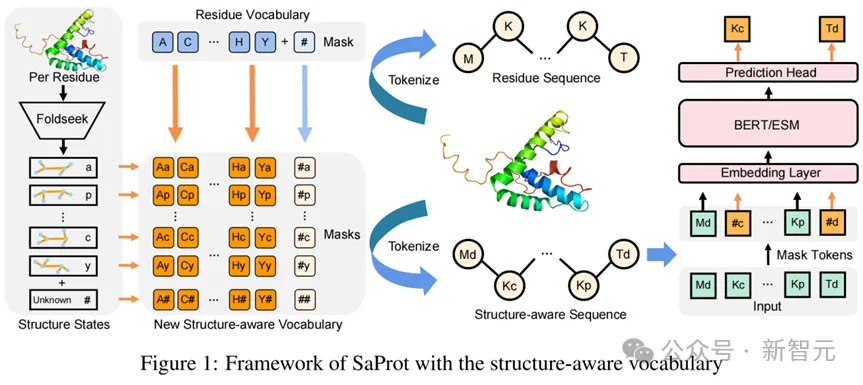
因此研究人员使用了一种简单而有效的结构嵌入方式:将结构词表和氨基酸词表计算笛卡尔积(即两两组合),形成新的结构感知词表。
这样对于蛋白质的每个位点,其氨基酸类型和对应的局部结构都能组合成新词表中的某个元素,从而让模型同时考虑到蛋白质的序列与结构信息。
本文使用Bert架构进行掩码语言建模(Masked Language Modeling )预训练(关于训练的更多细节请参考原论文)。
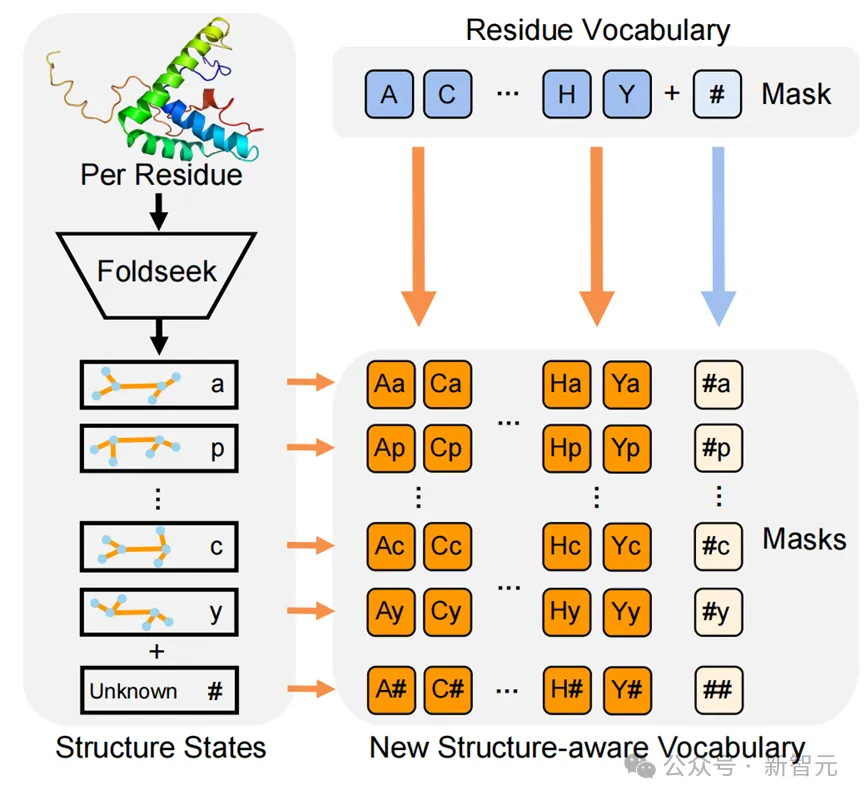
结构感知词表
实验
方法对比
一个可能令人疑惑的问题就是为什么需要这样编码结构?论文展示了使用不同的结构编码方式进行预训练的结果图:
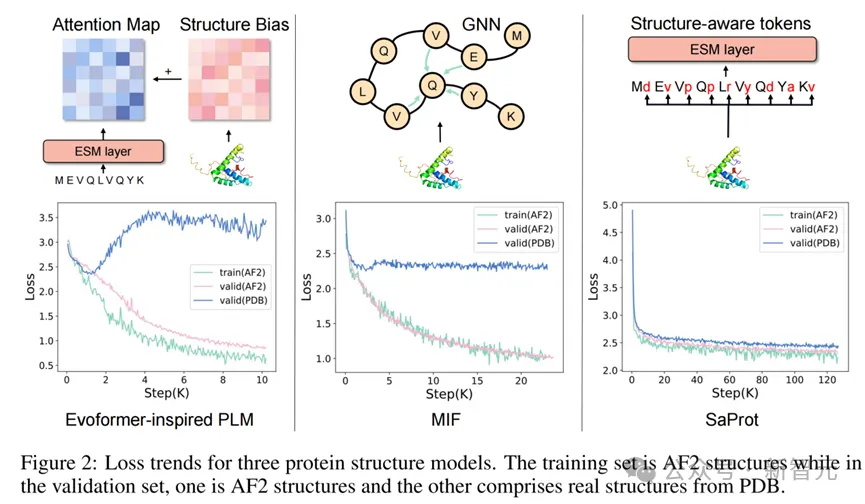
不同结构模型训练的loss曲线图
图左和图中是两种经典的蛋白质结构建模方式,即将结构信息编码成bias后添加到transformer的attention map中(如Evoformer,Uni-Mol),或者使用图神经网络的方式建模蛋白质的空间关系(如MIF,GearNet等)。
然而从loss图中可以发现,当上述两种建模方式在AF2结构上使用MLM的训练目标进行预训练时,模型会非常迅速地过拟合(表现为在AF2预测结构上预测loss非常低,但在PDB真实结构上loss停滞甚至上升)。
作者推测这是由于AF2预测出来的蛋白质结构带有一些隐藏的模式(patterns),由于前两种方式是直接对蛋白质的三维坐标进行建模,这些隐藏的pattern可能很轻易地就被模型识别出来,从而造成了信息泄露的问题,让模型无需真正学习到蛋白质的进化信息就能轻松地完成训练目标。
而结构感知词表通过将蛋白质结构编码成一维的结构序列,在尽可能保留结构模式的情况下忽略了精细的坐标数值,因此模型能够有效地利用结构信息而不受到隐藏pattern的影响。
Zero-shot测试
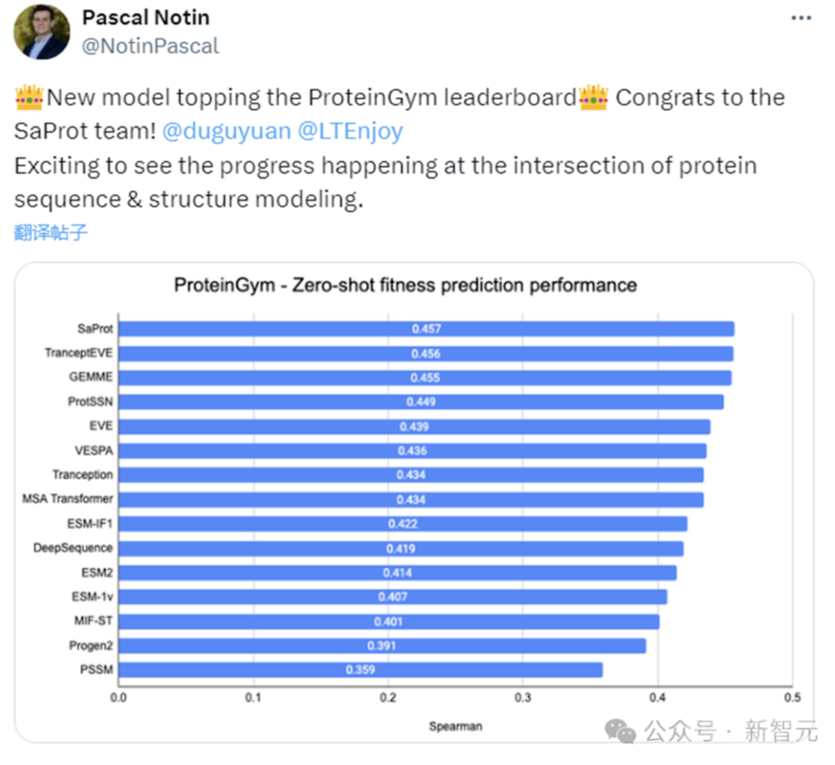
作者在蛋白质突变数据集(ProteinGym)上和真实人类临床疾病数据集(ClinVar)上测试了SaProt的zero-shot能力,结果如下:
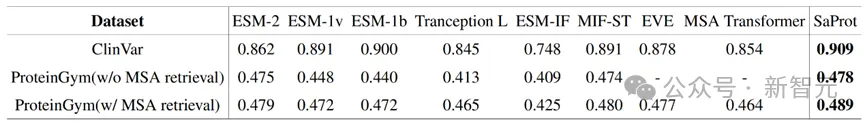
Zero-shot实验结果
SaProt在两个数据集上都超越了以往的所有结构和序列模型,证明了其在zero-shot预测突变上具备优异的能力。
监督微调测试
本文还涵盖了各种下游任务来测试模型表现,结果如下:
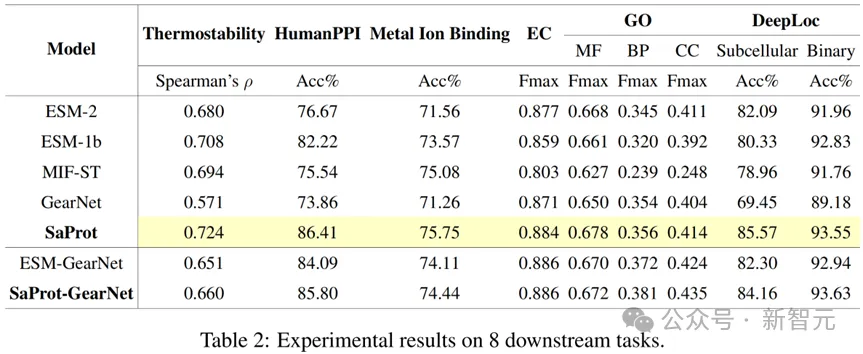
下游任务fine-tune结果
SaProt在各个下游任务上都超越了以往的序列和结构模型,展示出了其强大且通用的表征能力。
结构信息测试
SaProt在4000万的蛋白质结构上进行训练,获得了强大的表征能力。一个可能的疑问是如何确定SaProt学到了更多的结构信息而不是模型被训练得更好?论文对SaProt和ESM-2在残基接触预测任务(Contact Prediction Task)上进行了测试。作者冻住了模型的backbone,只训练一个线性分类层。
实验结果如下:
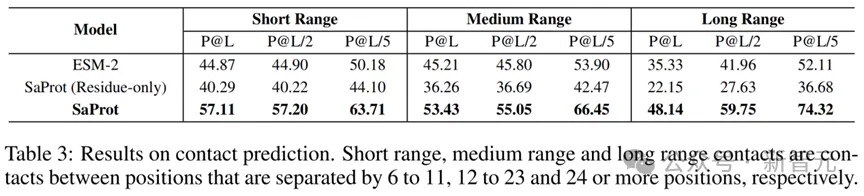
Contact Prediction Task的结果
从结果可以看到,由于结构token的嵌入,SaProt的表现大大超越了ESM-2,这表明SaProt蕴含了非常丰富的结构信息,使其能够在结构预测任务上获得十分优异的结果。
同时,论文在SCOPe数据库上对alpha蛋白质和beta蛋白质进行了可视化,结果如下:
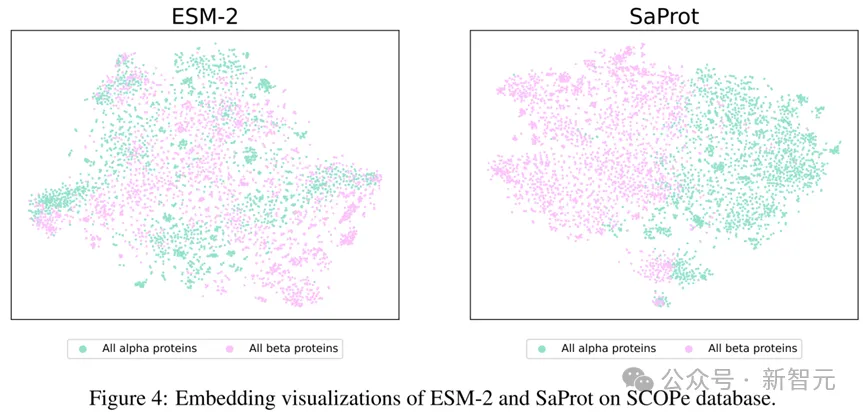
在SCOPe数据库上的Embedding可视化
SaProt的可视化结果非常清晰地将alpha蛋白质和beta蛋白质区分开来,而ESM-2的可视化结果却将两种蛋白质混杂在一起,这说明了SaProt对结构的变化有很强的感知能力。
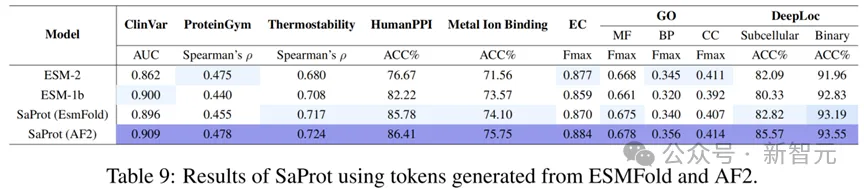
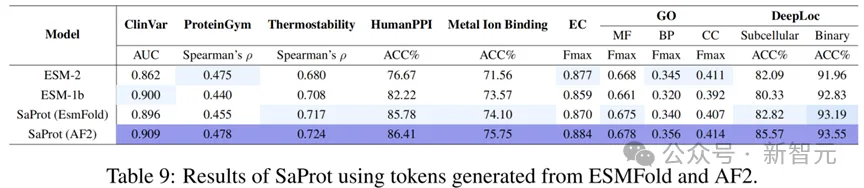
不同结构预测方法的比较
除了AF2,目前还存在许多其他的单序列结构预测方法(如ESMFold),因此本文额外测试了其他方法预测出来的结构对SaProt性能的作用。
结果如下:
不同结构预测方法的fine-tune结果
从测试结果可以看出,虽然SaProt在AF2结构上的表现非常好(模型本身也是基于AF2结构进行训练的),但其他的结构预测方法也能让SaProt与ESM-2等模型性能相当。
这意味着考虑到计算与时间成本,单序列结构预测模型也能作为替代方法输入到SaProt中。
局限
虽然SaProt经过训练展示出了优异的性能,但依然还有一些可以改进的地方,例如:
1. Foldseek默认的结构词表大小只有20,如果有更加精准的结构编码模型,扩大结构表征的词表大小,是不是能进一步提升模型利用结构的能力?
2. 由于计算能力的限制,SaProt只在650M上完成了训练。如果能够继续扩大模型规模,是否可以进一步地提升模型表现?
3. 论文虽然已经测试了很多的蛋白质任务,但还有一些其他任务可以应用探索,例如蛋白质序列设计(给定backbone预测氨基酸序列)等。




还没有评论,来说两句吧...